RT-PCR Protocol

Introduction
In 1987, Powell et al. advanced RT-PCR applications, combining reverse transcription with PCR amplification to convert RNA into complementary DNA (cDNA), which is then amplified with a thermostable DNA polymerase. This technique has greatly advanced molecular biology, enabling gene expression analysis, cDNA library construction, cloning, sequencing, and RNA characterization.
Principle
RT-PCR combines reverse transcription and PCR amplification to produce cDNA from RNA. Reverse transcriptase uses the RNA sequence as a template to synthesize single-stranded DNA, which then serves as the template for PCR. Primers targeting specific RNA coding regions enhance the reaction for a chosen transcript and are also used for cloning, while poly-dT oligos prime most RNAs by binding to their poly-A tails.
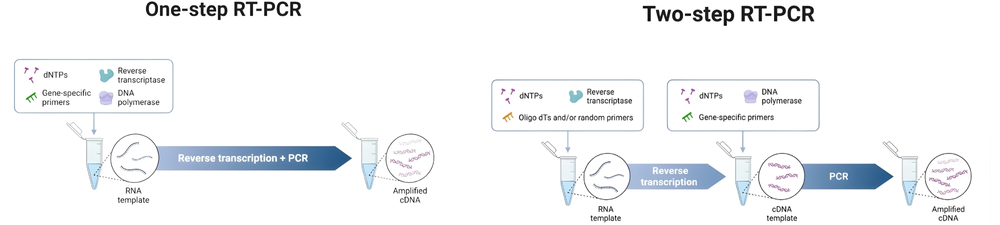
RT-PCR can be performed as either a one-step or two-step process. In one-step RT-PCR, both reverse transcription and PCR occur in the same tube using sequence-specific primers, which streamlines the workflow and reduces contamination risk. However, this method limits primer choice and is less flexible. Two-step RT-PCR separates the reactions into different tubes, allowing use of oligo(dT), random hexamer, or gene-specific primers during reverse transcription. This approach is more versatile and suitable for analyzing multiple genes from one RNA sample but involves more handling and higher contamination risk. One-step RT-PCR is preferred for high-throughput screening, while two-step RT-PCR is better for detailed, multi-target analysis.
Equipment, Reagents & Consumables
| A. Equipment | B. Reagents & Consumables | |
|
|
Protocol
I - RNA Extraction
Two RNA extraction methods are elaborated: one optimized for cells and tissues, and another designed specifically for vegetative yeast and tough meiotic spores.
A - Cells and Tissues RNA Extraction Method
Sample sizes: cells grown in culture (adherent or suspension) or small tissue samples (≤20 mg frozen tissue per 1 mL TRIzol)..
- Prepare workspace & samples
- Work in an RNase-free area: pre-cool centrifuges and have ice ready. Wear gloves and use filter tips.
- Aspirate / wash (cells)
- For adherent cells: remove media, wash once with ice-cold PBS. For suspension cells: pellet at 300 × g for 5 min and remove supernatant
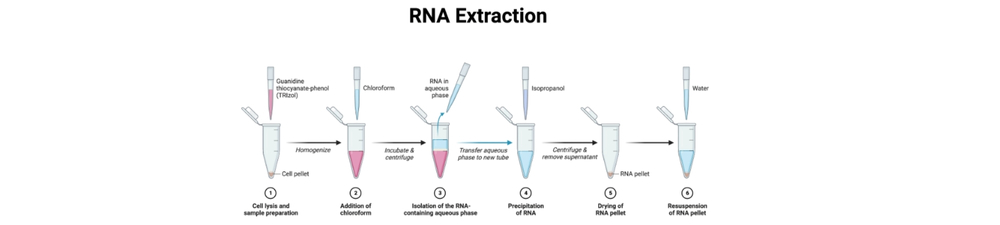
- Lyse in TRIzol
- Add 1 mL TRIzol per 10 cm² culture dish (or 1 mL per 50–100 mg tissue; for suspension cells add 0.75 mL TRIzol per 0.25 mL sample, per Chomczynski adaptation). Homogenize (pipetting or homogenizer). Incubate 5 min at room temperature.
- Separate using chloroform
- Add 0.2 mL chloroform per 1 mL TRIzol. Shake vigorously for 15 s; incubate 2–5 min at room temperature. Centrifuge at 12,000 × g for 15 min at 4 °C. After centrifugation, there will be three phases. Carefully transfer the clear upper aqueous phase (RNA) to a new RNase-free tube, leaving approximately 1 mm above the interphase to avoid DNA/phenol contamination.
- Precipitate RNA
- Add 0.5 mL isopropanol per 1 mL TRIzol, mix gently and incubate 5–10 min at room temperature. Centrifuge at 12,000 × g for 10 min at 4 °C to pellet RNA (increase time if expected yield is low).
- Wash pellet
- Remove supernatant and wash pellet with 1 mL 75% ethanol (prepared from 100% ethanol + RNase-free water). Vortex briefly to wash salts. Centrifuge at 7,500–11,500 × g for 5 min at 4 °C. Remove ethanol; air-dry briefly – do not overdry (2–5 min).
- Resuspend
- Resuspend RNA pellet in RNase-free water or TE (e.g., 15–50 µL depending on pellet size). Let dissolve on ice or at 55–65 °C briefly if needed. Measure concentration and purity (A260/A280, A260/A230) on NanoDrop.
- Optional DNase treatment
- If downstream RT-PCR shows genomic DNA contamination, treat with RNase-free DNase I (e.g., 1 U DNase I per 1–5 µg RNA, 37 °C for 10–15 min), then purify RNA away from DNase.
- Storage
- Short-term: −20 °C; long-term: −80 °C. Avoid repeated freeze-thaw cycles.
Quality Control & Tips
-
A260/A280 ratios of approximately 1.8–2.2 indicate acceptable nucleic acid purity; a low A260/A230 ratio suggests phenol or solvent contamination, warranting re-extraction.
- Run small aliquot on denaturing agarose gel or Bioanalyzer for integrity (rRNA bands, RIN).
- Work rapidly and on ice after lysis steps to minimize RNase activity.
- Handle TRIzol and phenol/chloroform under a fume hood.
- Collect phenolic waste separately in halogenated waste containers per institutional biosafety guidelines.
- Use glycogen carrier for low yields; prolong precipitation at −20 °C if necessary.
- Include a blank extraction (no input) to control for contamination in reagents.
B - Yeast and Tough Meiotic Spores RNA Extraction Methods
Two procedures are described: Procedure 1: hot PCA for vegetative yeast and Procedure 2: bead-beating + phenol for meiotic/tough cells.
Procedure 1: Hot PCA for Vegetative Yeast
Use aliquot ≤10 mL at A600=0.5 (avoid overloading Phase-Lock Gel). Pellet cells and remove media.
- Resuspend pellet in 400 µL Tris-EDTA buffer in 2 mL microcentrifuge tube.
- Add 40 µL 10% SDS + 400 µL PCA (25:24:1). Vortex and incubate at 65 °C for 10 min, vortex briefly every minute.
- Chill 5 min on ice; transfer to pre-spun Phase-Lock Gel tube (gel spun to bottom briefly). Centrifuge at 14,000 rpm for 5 min to separate phases.
- Wash aqueous phase with chloroform twice (400 µL each), centrifuge 14,000 rpm 5 min.
- Precipitate RNA: add 50 µL 3 M NaOAc pH 5.2 and bring to final 2 mL with 100% ethanol; invert mix – centrifuge at 14,000 rpm for 5–10 min.
- Wash pellet with 70% ethanol, centrifuge, air-dry briefly, resuspend in RNase-free water.
Procedure 2: Bead-Beating + Phenol for Meiotic/Tough Cells
- Pellet ≤10 mL culture at A600=0.5; resuspend in 0.5 mL lysis buffer; add 1 mL water-saturated phenol and acid-washed glass beads.
- Vortex cycles: 20 s vortex → 20 s ice slurry, repeat cycles for total approximately 7 min (protect sample from heating).
- Add extra phenol, lysis buffer, and 200 µL 10% SDS; vortex more, centrifuge 4000 × g 10 min at 4 °C.
- Transfer aqueous to new tube, extract with PCA (25:24:1) as above, then precipitate with 2.5 volumes 100% ethanol at −20 °C ≥90 min. Centrifuge at 14,000 rpm for 10 min.
- Wash with 70% ethanol, resuspend; if processing multiple aliquots, combine pellets, then follow Procedure 1 cleanup steps.
Quality Control & Tips
- Check A270 vs A260 to detect phenol contamination (if A270 > A260 → phenol present)
- Ensure complete cell breakage – incomplete lysis biases the RNA population (tRNA:rRNA ratios change)
N.B. Although the above procedures describe reagent-based, kit-free RNA extraction methods, RNA extraction can also be performed using commercial RNA extraction kits.
II - Reverse Transcription
Reverse transcription is a critical step in RT-PCR, converting RNA into complementary DNA (cDNA) for subsequent amplification and analysis.
Denaturation / Primer Annealing Mix (10 µL)
-
In RNase-free tube combine: RNA (typically 0.1–1 µg), 1 µL 50 µM oligo(dT) (or 1 µL random hexamers / gene-specific), 1 µL 10 mM dNTPs, and RNase-free water to 10 µL. Mix gently.
- Heat-denature
- Incubate at 65 °C for 5 minutes to reduce RNA secondary structure; then place on ice (brief spin).
- Prepare RT master mix (10 µL per reaction) on ice (scale for number of reactions +10%): per reaction include: 2 µL 10× RT buffer, 4 µL 25 mM MgCl₂ (if needed), 2 µL 0.1 M DTT (if required), 1 µL RNase inhibitor, 1 µL reverse transcriptase. Keep enzyme last and keep mix on ice.
- Assemble reaction
- Add 10 µL RT master mix to each denatured 10 µL RNA/primer tube → 20 µL final. Mix gently, spin briefly. Include a No-reverse transcriptase control (omit enzyme, replace with equal volume of buffer) for each RNA sample.
- Incubation (cDNA synthesis)
- Incubate at 50 °C for 50 minutes.
Note: Classic reverse transcriptase may use 37–42 °C; thermostable reverse transcriptases allow 50–55 °C to help with structured/GC-rich templates.
- Terminate reverse transcription / inactivate enzyme
- Heat to 85 °C for 5 minutes, then cool on ice. Brief spin.
- Optional RNase H treatment
- Add 1 µL RNase H, incubate 37 °C for 20 min to degrade RNA in RNA:cDNA hybrids (improves downstream PCR sometimes). Then proceed to PCR or store cDNA.
- Storage
- Store cDNA at −20 °C (short-term) or −80 °C (long term). Avoid freeze/thaw cycles.
Quality Control & Tips
-
RNase-free technique & reagent handling: Reverse transcription reactions are highly sensitive to RNase and reagent quality. Prepare reactions in a dedicated RNase-free hood/area, wear gloves, use filtered tips, and aliquot enzyme/dNTP stocks to avoid repeated freeze–thaw. Keep reverse transcriptase enzyme on ice during setup and add last to the master mix.
-
Primer choice & its effect on products: Primer selection determines cDNA representation. Oligo(dT) primers target polyadenylated mRNA (with a 3′ bias) and are suitable when full-length mRNA representation or mRNA-specific cDNA is desired. Random hexamers prime broadly, including non-polyadenylated RNAs, and are used when even coverage or small RNAs are needed. Gene-specific primers provide the highest specificity and are mandatory for one-step RT-PCR assays.
-
Enzyme & temperature considerations: Thermostable reverse transcriptases tolerate higher temperatures (50–55 °C), helping read through structured templates. If your target is GC-rich or highly structured, consider a thermostable reverse transcriptase and higher incubation temperature within enzyme specs. Always consult the enzyme datasheet for optimum Mg²⁺ and temperature.
-
Reverse transcription efficiency & verification: To verify reverse transcription worked, amplify a short fragment of a housekeeping gene (ACTB, GAPDH) from your cDNA; consistent Ct values or clear electrophoresis bands across technical replicates indicate good reverse transcription efficiency. If you need quantitative measures of reverse transcriptase efficiency, use spike-in controls (e.g., synthetic RNA standards of known concentration).
-
DNase treatment & No-reverse transcriptase controls: Always include a No-reverse transcriptase control for each sample to detect genomic DNA contamination. If a No-reverse transcriptase control gives a PCR product, re-treat RNA with RNase-free DNase I and repeat reverse transcription.
III - Obtaining & Analyzing the RT-PCR Endpoint Product
Endpoint PCR amplifies the cDNA generated from reverse transcription, enabling the detection and analysis of specific cDNA sequences corresponding to target RNA.
Endpoint PCR Setup (25 µL Example)
- Reaction mix (25 µL):
- 12.5 µL 2× PCR master mix (hot-start Taq or high-fidelity mix).
- 0.5 µL forward primer (10 µM) (final 0.2 µM).
- 0.5 µL reverse primer (10 µM).
- 1–2 µL cDNA template (dilute reverse transcription product 1:5 or 1:10 if inhibitors suspected).
- Nuclease-free water to 25 µL.
- Controls (include in every run):
- No-template control (NTC) – water instead of cDNA.
- No-reverse transcriptase control – PCR using the negative reverse transcriptase sample to detect genomic DNA.
- Positive control – template known to amplify (plasmid, previously validated cDNA)
- Cycling conditions (typical) – adjust to primer Tm (melting temperature, calculated from primer sequence) & polymerase:
-
Initial denaturation / enzyme activation
-
95 °C for 2–3 minutes (hot-start activation if required).
-
-
30–35 cycles of:
-
Denaturation: 95 °C for 15–30 seconds.
-
Annealing: (primer Tm − 3 to −5 °C) for 15–30 seconds.
-
Extension: 72 °C for 30 seconds to 1 minute per kilobase (kb) of expected product.
-
-
Final extension: 72 °C for 5–7 minutes.
-
Hold: 4 °C (or remove samples).
-
Agarose Gel Electrophoresis: Steps & Analysis
-
Gel selection & preparation: Choose agarose percentage by amplicon size (1% general; 1.5–2% for 100–500 base pairs). Prepare gel in 1× TE buffer with staining dye per manufacturer instructions.
- Loading: Mix 5–10 µL PCR product with loading dye; load ladder and samples.
- Run conditions: 5–8 V/cm (e.g., 100 V for standard mini-gel) until separation is adequate (30–60 min). Visualize and image on blue-light/UV transilluminator.
- Interpretation (detailed):
- Single band at expected size (clean): Specific amplification; good product for sequencing or cloning. Check intensity relative to ladder – strong band indicates good yield.
- No band (but positive control OK): Indicates reverse transcription or PCR failure for that sample – possible causes: degraded RNA, inhibitors, wrong primer/dilution. Try increasing template, re-do reverse transwcription, or redesign primers.
- Band in No-reverse transcriptase control: Genomic DNA contamination – re-treat RNA with DNase I and repeat reverse transcription; redesign primers to span exon–exon junctions if possible.
- Multiple bands / smears: Nonspecific priming or alternative splice variants. Remedies: raise annealing temperature, reduce cycles, use hot-start polymerase, redesign primers, or gel-purify single band for downstream use.
Recommendations for Sanger Sequencing & Cloning
Sequencing: Use high-fidelity polymerase for PCR if planning Sanger sequencing (reduces polymerase errors). Provide purified PCR product at provider-specified concentration (commonly 10–20 ng/µL for short amplicons) and the sequencing primer (forward ± reverse if product >800–1000 bp). If sequencing shows mixed peaks, suspect mixed templates – consider cloning.
Cloning: For TA cloning use Taq-amplified products (A-overhangs). For blunt cloning (high-fidelity enzymes) use appropriate blunt ligation or add A-tails enzymatically. Sequence plasmid clones to confirm insert identity and to check for PCR-introduced mutations.
IV - Interpretation
-
The reaction containing reverse transcriptase produces the expected amplification band, whereas the reaction without reverse transcriptase shows no band.
→ Interpretation: Successful reverse transcription from RNA; the amplified product originates from complementary DNA (cDNA) synthesized from RNA, not from contaminating genomic DNA.
→ Action: Proceed to product purification, sequencing, or other downstream analyses. - Both the reaction with reverse transcriptase and the reaction without reverse transcriptase produce amplification bands of identical size.
→ Interpretation: The RNA sample is contaminated with genomic DNA.
→ Action: Treat the RNA with DNase I to remove residual DNA, re-purify the RNA, and repeat the reverse transcription.
For future assays, design primers spanning exon–exon junctions to ensure amplification only from processed messenger RNA (mRNA) rather than genomic DNA. - No amplification product is detected in the reaction containing reverse transcriptase, but the positive control amplifies successfully.
→ Interpretation: The problem is specific to the tested RNA sample. Possible causes include degraded RNA, presence of PCR inhibitors, inefficient priming, or reverse transcriptase failure.
→ Action: Evaluate RNA integrity by running a small aliquot on an agarose gel or using a Bioanalyzer, and assess purity using A₂₆₀/A₂₈₀ ratios. If RNA quality is poor, re-extract the RNA.
Consider modifying reverse transcription conditions by using a different primer type (e.g., random hexamers instead of oligo(dT) primers), increasing RNA input, or extending the reverse transcription incubation time. - Multiple amplification bands are observed.
→ Interpretation: Non-specific amplification occurred.
→ Action: Verify primer specificity by in-silico analysis, increase the annealing temperature, and use a hot-start DNA polymerase to reduce non-specific priming.
If alternative splice variants are suspected, excise individual bands from the gel, purify them, and sequence each to confirm transcript identity.
Avoid nested PCR unless necessary, as it increases the risk of contamination. - Smearing of amplification products is observed on the gel.
→ Interpretation: The RNA template is degraded, or gel wells were overloaded.
→ Action: Improve RNA integrity through careful handling and immediate stabilization after extraction, reduce template loading, and confirm that electrophoresis conditions are appropriate. If necessary, repeat the reaction with purified template and optimized cycle number. - Amplification yields are low or faint bands are visible.
→ Interpretation: Low initial RNA concentration, inefficient reverse transcription, or presence of PCR inhibitors (e.g., residual phenol or ethanol) may be responsible.
→ Action: Measure RNA concentration and purity using A₂₆₀/A₂₃₀ ratios—low values indicate contamination with phenol or guanidinium compounds. Ensure RNA pellets are properly dried before resuspension. If inhibitors persist, re-extract the RNA or perform additional ethanol washes before repeating the RT-PCR.
Conclusion
This protocol provides a robust framework for RNA extraction, reverse transcription, and endpoint RT-PCR, enabling reliable gene expression analysis, cloning, and sequencing. By following these steps and quality control measures, researchers can achieve high-quality cDNA and PCR products for downstream applications in molecular biology.
References:
-
Isolation of Total RNA from Yeast Cell Cultures by Manuel Ares, published in Cold Spring Harbor Protocols, 2012.
-
Reverse-Transcription PCR (RT-PCR) by Julia Bachman, Department of Neuroscience, Johns Hopkins University School of Medicine, published by Elsevier Inc., 2013.
-
The Polymerase Chain Reaction (PCR), qPCR, and RT-PCR by Mehdi Jalali, Justyna Zaborowska, and Morteza Jalali, University of Liverpool and University of Oxford, published by Elsevier Inc., 2017.