Protocole de RT-PCR

Introduction
En 1987, Powell et al. ont fait progresser les applications de la RT-PCR, en combinant la transcription inverse avec l’amplification par PCR pour convertir l’ARN en ADN complémentaire (cDNA), qui est ensuite amplifié à l’aide d’une ADN polymérase thermostable. Cette technique a considérablement avancé la biologie moléculaire, permettant l’analyse de l’expression génique, la construction de bibliothèques de cDNA, le clonage, le séquençage et la caractérisation de l’ARN.
Principe
La RT-PCR combine la transcription inverse et l’amplification par PCR pour produire de l’cDNA à partir de l’ARN. La transcriptase inverse utilise la séquence d’ARN comme matrice pour synthétiser de l’ADN simple brin, qui sert ensuite de matrice pour la PCR. Des amorces ciblant des régions codantes spécifiques de l’ARN améliorent la réaction pour un transcrit choisi et sont également utilisées pour le clonage, tandis que les oligomères poly-dT amorcent la plupart des ARN en se fixant à leurs queues poly-A.
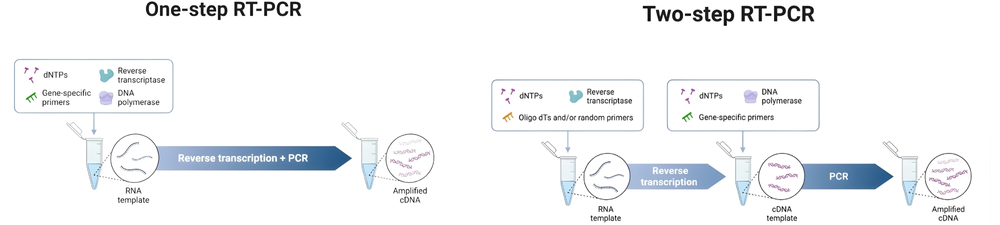
La RT-PCR peut être réalisée en une ou deux étapes. Dans la RT-PCR en une étape, la transcription inverse et la PCR se déroulent dans le même tube avec des amorces spécifiques à la séquence, ce qui simplifie le flux de travail et réduit le risque de contamination. Cependant, cette méthode limite le choix des amorces et est moins flexible. La RT-PCR en deux étapes sépare les réactions dans différents tubes, permettant l’utilisation d’oligo(dT), d’hexamères aléatoires ou d’amorces spécifiques à un gène pendant la transcription inverse. Cette approche est plus polyvalente et adaptée à l’analyse de plusieurs gènes à partir d’un même échantillon d’ARN, mais implique plus de manipulations et un risque de contamination plus élevé. La RT-PCR en une étape est préférée pour le criblage à haut débit, tandis que la RT-PCR en deux étapes est meilleure pour une analyse détaillée de cibles multiples.
Équipements, Réactifs & Consommables
| A. Équipements | B. Réactifs & Consommables | |
|
|
Protocole
I - Extraction d’ARN
Deux méthodes d’extraction d’ARN sont décrites : l’une optimisée pour les cellules et les tissus, et l’autre conçue spécifiquement pour les levures végétatives et les spores méiotiques résistantes.
A - Méthode d’extraction d’ARN pour cellules et tissus
Tailles des échantillons : cellules cultivées en culture (adhérentes ou en suspension) ou petits échantillons de tissu (≤20 mg de tissu congelé par 1 mL de TRIzol).
- Préparer l’espace de travail et les échantillons
- Travailler dans une zone exempte de RNase : pré-refroidir les centrifugeuses et préparer de la glace. Porter des gants et utiliser des embouts à filtre.
- Aspirer / laver (cellules)
- Pour les cellules adhérentes : retirer le milieu, laver une fois avec du PBS glacé. Pour les cellules en suspension : centrifuger à 300 × g pendant 5 min et retirer le surnageant.
- Lyser dans TRIzol
- Ajouter 1 mL de TRIzol par 10 cm² de boîte de culture (ou 1 mL par 50–100 mg de tissu ; pour les cellules en suspension, ajouter 0,75 mL de TRIzol par 0,25 mL d’échantillon, selon l’adaptation de Chomczynski). Homogénéiser (pipetage ou homogénéisateur). Incuber 5 min à température ambiante.
- Séparer avec du chloroforme
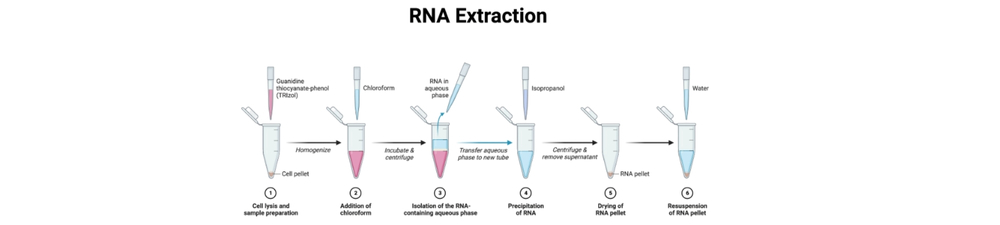
- Ajouter 0,2 mL de chloroforme par 1 mL de TRIzol. Agiter vigoureusement pendant 15 s ; incuber 2–5 min à température ambiante. Centrifuger à 12 000 × g pendant 15 min à 4 °C. Après centrifugation, trois phases apparaîtront. Transférer soigneusement la phase aqueuse supérieure claire (ARN) dans un nouveau tube exempt de RNase, en laissant environ 1 mm au-dessus de l’interphase pour éviter la contamination par l’ADN/phénol.
- Précipiter l’ARN
- Ajouter 0,5 mL d’isopropanol par 1 mL de TRIzol, mélanger doucement et incuber 5–10 min à température ambiante. Centrifuger à 12 000 × g pendant 10 min à 4 °C pour précipiter l’ARN (augmenter le temps si le rendement attendu est faible).
- Laver le culot
- Retirer le surnageant et laver le culot avec 1 mL d’éthanol à 75 % (préparé à partir d’éthanol 100 % + eau exempte de RNase). Vortexer brièvement pour éliminer les sels. Centrifuger à 7 500–11 500 × g pendant 5 min à 4 °C. Retirer l’éthanol ; sécher à l’air brièvement – ne pas trop sécher (2–5 min).
- Resuspendre
- Resuspendre le culot d’ARN dans de l’eau exempte de RNase ou du TE (par ex., 15–50 µL selon la taille du culot). Laisser dissoudre sur glace ou à 55–65 °C brièvement si nécessaire. Mesurer la concentration et la pureté (A260/A280, A260/A230) avec un NanoDrop.
- Traitement optionnel à la DNase
- Si la RT-PCR en aval montre une contamination par l’ADN génomique, traiter avec de la DNase I exempte de RNase (par ex., 1 U de DNase I par 1–5 µg d’ARN, 37 °C pendant 10–15 min), puis purifier l’ARN pour éliminer la DNase.
- Stockage
- Court terme : −20 °C ; long terme : −80 °C. Éviter les cycles de congélation/décongélation répétés.
Contrôle de qualité & Conseils
-
Les ratios A260/A280 d’environ 1,8–2,2 indiquent une pureté acceptable des acides nucléiques ; un faible ratio A260/A230 suggère une contamination par du phénol ou des solvants, nécessitant une ré-extraction.
- Analyser un petit aliquot sur un gel d’agarose dénaturant ou un Bioanalyzer pour vérifier l’intégrité (bandes d’ARNr, RIN).
- Procéder rapidement et maintenir les échantillons sur glace après les étapes de lyse afin de réduire l’activité des RNases.
- Manipuler le TRIzol et le phénol/chloroforme sous une hotte à flux laminaire.
- Collecter les déchets phénoliques séparément dans des conteneurs pour déchets halogénés conformément aux directives de biosécurité institutionnelles.
- Utiliser un porteur de glycogène pour les faibles rendements ; prolonger la précipitation à −20 °C si nécessaire.
- Inclure une extraction à blanc (sans échantillon) pour contrôler la contamination des réactifs.
B - Méthodes d’extraction d’ARN pour levures et spores méiotiques résistantes
Deux procédures sont décrites : Procédure 1 : PCA chaud pour les levures végétatives et Procédure 2 : broyage à billes + phénol pour les cellules méiotiques/résistantes.
Procédure 1 : PCA chaud pour les levures végétatives
Utiliser un aliquot ≤10 mL à A600=0,5 (éviter de surcharger le gel de séparation de phase). Centrifuger les cellules et retirer le milieu.
- Resuspendre le culot dans 400 µL de tampon Tris-EDTA dans un tube de microcentrifugation de 2 mL.
- Ajouter 40 µL de SDS à 10 % + 400 µL de PCA (25:24:1). Vortexer et incuber à 65 °C pendant 10 min, vortexer brièvement toutes les minutes.
- Refroidir 5 min sur glace ; transférer dans un tube à gel de séparation de phase pré-centrifugé (gel au fond brièvement). Centrifuger à 14 000 rpm pendant 5 min pour séparer les phases.
- Laver la phase aqueuse avec du chloroforme deux fois (400 µL à chaque fois), centrifuger à 14 000 rpm pendant 5 min.
- Précipiter l’ARN : ajouter 50 µL d’acétate de sodium 3 M pH 5,2 et porter à un volume final de 2 mL avec de l’éthanol 100 % ; mélanger par inversion – centrifuger à 14 000 rpm pendant 5–10 min.
- Laver le culot avec de l’éthanol à 70 %, centrifuger, sécher à l’air brièvement, resuspendre dans de l’eau exempte de RNase.
Procédure 2 : Broyage à billes + phénol pour cellules méiotiques/résistantes
- Centrifuger ≤10 mL de culture à A600=0,5 ; resuspendre dans 0,5 mL de tampon de lyse ; ajouter 1 mL de phénol saturé en eau et des billes de verre lavées à l’acide.
- Cycles de vortex : vortexer 20 s → 20 s dans un bain de glace, répéter les cycles pendant environ 7 min au total (protéger l’échantillon de la chauffe).
- Ajouter du phénol supplémentaire, du tampon de lyse et 200 µL de SDS à 10 % ; vortexer davantage, centrifuger à 4000 × g pendant 10 min à 4 °C.
- Transférer la phase aqueuse dans un nouveau tube, extraire avec du PCA (25:24:1) comme ci-dessus, puis précipiter avec 2,5 volumes d’éthanol 100 % à −20 °C pendant ≥90 min. Centrifuger à 14 000 rpm pendant 10 min.
- Laver avec de l’éthanol à 70 %, resuspendre ; si plusieurs aliquots sont traités, combiner les culots, puis suivre les étapes de nettoyage de la Procédure 1.
Contrôle de qualité & Conseils
- Vérifier A270 vs A260 pour détecter une contamination par le phénol (si A270 > A260 → phénol présent).
- S’assurer d’une rupture complète des cellules – une lyse incomplète biaise la population d’ARN (les ratios tRNA:ARNr changent).
N.B. Bien que les procédures ci-dessus décrivent des méthodes d’extraction d’ARN sans kit basées sur des réactifs, l’extraction d’ARN peut également être réalisée à l’aide de kits d’extraction d’ARN commerciaux.
II - Transcription inverse
La transcription inverse est une étape cruciale de la RT-PCR, convertissant l’ARN en ADN complémentaire (cDNA) pour une amplification et une analyse ultérieures.
Mélange de dénaturation / appariement des amorces (10 µL)
-
Dans un tube exempt de RNase, combiner : ARN (généralement 0,1–1 µg), 1 µL d’oligo(dT) 50 µM (ou 1 µL d’hexamères aléatoires / spécifique à un gène), 1 µL de dNTPs 10 mM, et de l’eau exempte de RNase jusqu’à 10 µL. Mélanger doucement.
- Dénaturation thermique
- Incuber à 65 °C pendant 5 minutes pour réduire la structure secondaire de l’ARN ; puis placer sur glace (centrifugation brève).
- Préparer le RT master mix (10 µL par réaction) sur glace (+10 % du nombre de réactions) : par réaction, inclure : 2 µL 10× du tampon de transcriptase inverse, 4 µL de MgCl₂ 25 mM (si nécessaire), 2 µL de DTT 0,1 M (si requis), 1 µL d’inhibiteur de RNase, 1 µL de transcriptase inverse. Conserver l’enzyme en dernier et maintenir le mélange sur glace.
- Assembler la réaction
- Ajouter 10 µL du RT master mix à chaque tube d’ARN/amorces dénaturé de 10 µL → 20 µL final. Mélanger doucement, centrifuger brièvement. Inclure un contrôle sans transcriptase inverse (omettre l’enzyme, remplacer par un volume équivalent de tampon) pour chaque échantillon d’ARN.
- Incubation (synthèse d’cDNA)
- Incuber à 50 °C pendant 50 minutes.
Note : La transcriptase inverse classique peut utiliser 37–42 °C ; les transcriptases inverses thermostables permettent 50–55 °C pour aider avec les matrices structurées/riches en GC.
- Terminer la transcription inverse / inactiver l’enzyme
- Chauffer à 85 °C pendant 5 minutes, puis refroidir sur glace. Centrifugation brève.
- Traitement optionnel à la RNase H
- Ajouter 1 µL de RNase H, incuber à 37 °C pendant 20 min pour dégrader l’ARN dans les hybrides ARN:cDNA (améliore parfois la PCR en aval). Ensuite, procéder à la PCR ou stocker l’cDNA.
- Stockage
- Stocker l’cDNA à −20 °C (court terme) ou −80 °C (long terme). Éviter les cycles de congélation/décongélation.
Contrôle de qualité & Conseils
-
Technique exempte de RNase & manipulation des réactifs : Les réactions de transcription inverse sont très sensibles aux RNases et à la qualité des réactifs. Préparer les réactions dans une hotte/zone dédiée exempte de RNase, porter des gants, utiliser des embouts à filtre et aliquoter les stocks d’enzymes/dNTP pour éviter les cycles de congélation-décongélation répétés. Conserver l’enzyme transcriptase inverse sur glace pendant la préparation et l’ajouter en dernier au master mix.
-
Choix des amorces & leur effet sur les produits : La sélection des amorces détermine la représentation de l’cDNA. Les amorces oligo(dT) ciblent les ARNm polyadénylés (avec un biais 3’) et sont adaptées lorsque la représentation complète de l’ARNm ou un cDNA spécifique à l’ARNm est souhaitée. Les hexamères aléatoires amorcent de manière large, y compris les ARN non polyadénylés, et sont utilisés lorsque une couverture uniforme ou des petits ARN sont nécessaires. Les amorces spécifiques à un gène offrent la plus grande spécificité et sont obligatoires pour les essais RT-PCR en une étape.
-
Considérations sur l’enzyme & la température : Les transcriptases inverses thermostables tolèrent des températures plus élevées (50–55 °C), facilitant la lecture à travers des matrices structurées. Si votre cible est riche en GC ou très structurée, envisager une transcriptase inverse thermostable et une température d’incubation plus élevée dans les spécifications de l’enzyme. Toujours consulter la fiche technique de l’enzyme pour un Mg²⁺ et une température optimaux.
-
Efficacité & vérification de la transcription inverse : Pour vérifier que la transcription inverse a fonctionné, amplifier un court fragment d’un gène de ménage (ACTB, GAPDH) à partir de votre cDNA ; des valeurs Ct cohérentes ou des bandes claires à l’électrophorèse dans les réplicats techniques indiquent une bonne efficacité de transcription inverse. Si des mesures quantitatives de l’efficacité de la transcription inverse sont nécessaires, utiliser des spike-in contrôles (par ex., standards d’ARN synthétiques de concentration connue).
-
Traitement à la DNase & contrôles sans transcriptase inverse : Toujours inclure un contrôle sans transcriptase inverse pour chaque échantillon afin de détecter une contamination par l’ADN génomique. Si un contrôle sans transcriptase inverse donne un produit PCR, retraiter l’ARN avec de la DNase I exempte de RNase et répéter la transcription inverse.
III - Obtention & Analyse du produit final RT-PCR
La PCR en point final amplifie l’cDNA généré par la transcription inverse, permettant la détection et l’analyse de séquences d’cDNA spécifiques correspondant à l’ARN cible.
Configuration de la PCR en point final (exemple de 25 µL)
- Mélange de réaction (25 µL) :
- 12,5 µL du master mix PCR 2× (Taq à démarrage à chaud ou mélange haute fidélité).
- 0,5 µL d’amorce sens (10 µM) (final 0,2 µM).
- 0,5 µL d’amorce antisens (10 µM).
- 1–2 µL de matrice d’cDNA (diluer le produit RT 1:5 ou 1:10 si des inhibiteurs sont suspectés).
- Eau exempte de nucléases jusqu’à 25 µL.
- Contrôles (inclure dans chaque série) :
- Contrôle sans matrice (NTC) – eau au lieu d’cDNA.
- Contrôle sans transcriptase inverse – PCR utilisant l’échantillon négatif de transcriptase inverse pour détecter l’ADN génomique.
- Contrôle positif – matrice connue pour s’amplifier (plasmide, cDNA précédemment validé).
- Conditions de cyclage (typiques) – ajuster à la Tm des amorces (température de fusion, calculée à partir de la séquence des amorces) & polymérase :
-
Dénaturation initiale / activation enzymatique
-
95 °C pendant 2–3 minutes (activation à chaud si requis).
-
-
30–35 cycles de :
-
Dénaturation : 95 °C pendant 15–30 secondes.
-
Appariement : (Tm des amorces − 3 à −5 °C) pendant 15–30 secondes.
-
Extension : 72 °C pendant 30 secondes à 1 minute par kilobase (kb) de produit attendu.
-
-
Extension finale : 72 °C pendant 5–7 minutes.
-
Maintien : 4 °C (ou retirer les échantillons).
-
Électrophorèse sur gel d’agarose : Étapes & Analyse
-
Sélection & préparation du gel : Choisir le pourcentage d’agarose selon la taille de l’amplicon (1 % général ; 1,5–2 % pour 100–500 paires de bases). Préparer le gel dans un tampon TE 1× avec un colorant de marquage selon les instructions du fabricant.
- Chargement : Mélanger 5–10 µL de produit PCR avec un colorant de charge ; charger une échelle et les échantillons.
- Conditions de migration : 5–8 V/cm (par ex., 100 V pour un mini-gel standard) jusqu’à ce que la séparation soit adéquate (30–60 min). Visualiser et imager sur un transilluminateur à lumière bleue/UV.
- Interprétation (détaillée) :
- Bande unique à la taille attendue (propre) : Amplification spécifique ; bon produit pour le séquençage ou le clonage. Vérifier l’intensité par rapport à l’échelle – une bande forte indique un bon rendement.
- Aucune bande (mais contrôle positif OK) : Indique un échec de la transcription inverse ou de la PCR pour cet échantillon – causes possibles : ARN dégradé, inhibiteurs, mauvaise amorce/dilution. Essayer d’augmenter la matrice, refaire la transcription inverse ou redessiner les amorces.
- Bande dans le contrôle sans transcriptase inverse : Contamination par l’ADN génomique – retraiter l’ARN avec de la DNase I et répéter la transcription inverse ; redessiner les amorces pour couvrir les jonctions exon-exon si possible.
- Bandes multiples / étalements : Amorcage non spécifique ou variantes d’épissage alternatives. Remèdes : augmenter la température d’hybridation, réduire le nombre de cycles, utiliser une polymérase à démarrage à chaud, redessiner les amorces ou purifier la bande unique sur gel pour une utilisation en aval.
Recommandations pour le séquençage Sanger & Clonage
Séquençage : Utiliser une polymérase haute fidélité pour la PCR si un séquençage Sanger est prévu (réduit les erreurs de polymérase). Fournir un produit PCR purifié à la concentration spécifiée par le prestataire (généralement 10–20 ng/µL pour les amplicons courts) et l’amorce de séquençage (sens ± antisens si le produit >800–1000 pb). Si le séquençage montre des pics mixtes, suspecter des matrices mixtes – envisager le clonage.
Clonage : Pour le clonage TA, utiliser des produits amplifiés par Taq (surplombs A). Pour le clonage blunt (enzymes haute fidélité), utiliser une ligation blunt appropriée ou ajouter des queues A par voie enzymatique. Séquencer les clones plasmidiques pour confirmer l’identité de l’insert et vérifier les mutations introduites par la PCR.
IV - Interprétation
-
La réaction contenant la transcriptase inverse produit la bande d’amplification attendue, tandis que la réaction sans transcriptase inverse ne montre aucune bande.
→ Interprétation : Transcription inverse réussie à partir de l’ARN ; le produit amplifié provient de l’ADN complémentaire (cDNA) synthétisé à partir de l’ARN, et non de l’ADN génomique contaminant.
→ Action : Procéder à la purification du produit, au séquençage ou à d’autres analyses en aval. - La réaction avec et sans transcriptase inverse produit des bandes d’amplification de taille identique.
→ Interprétation : L’échantillon d’ARN est contaminé par de l’ADN génomique.
→ Action : Traiter l’ARN avec de la DNase I pour éliminer l’ADN résiduel, repurifier l’ARN et répéter la transcription inverse.
Pour les futurs essais, concevoir des amorces couvrant les jonctions exon-exon pour garantir l’amplification uniquement à partir de l’ARNm traité plutôt que de l’ADN génomique. - Aucun produit d’amplification n’est détecté dans la réaction contenant la transcriptase inverse, mais le contrôle positif s’amplifie avec succès.
→ Interprétation : Le problème est spécifique à l’échantillon d’ARN testé. Les causes possibles incluent un ARN dégradé, la présence d’inhibiteurs de PCR, un amorçage inefficace ou un échec de la transcriptase inverse.
→ Action : Évaluer l’intégrité de l’ARN en analysant un petit aliquot sur un gel d’agarose ou avec un Bioanalyzer, et évaluer la pureté à l’aide des ratios A₂₆₀/A₂₈₀. Si la qualité de l’ARN est médiocre, ré-extraire l’ARN.
Envisager de modifier les conditions de transcription inverse en utilisant un type d’amorce différent (par ex., hexamères aléatoires au lieu d’amorces oligo(dT)), augmenter l’apport d’ARN ou prolonger le temps d’incubation de la transcription inverse. - Plusieurs bandes d’amplification sont observées.
→ Interprétation : Une amplification non spécifique s’est produite.
→ Action : Vérifier la spécificité des amorces par analyse in silico, augmenter la température d’hybridation et utiliser une ADN polymérase à démarrage à chaud pour réduire l’amorçage non spécifique.
Si des variantes d’épissage alternatives sont suspectées, exciser les bandes individuelles du gel, les purifier et séquencer chacune pour confirmer l’identité du transcrit.
Éviter la PCR nichée sauf si nécessaire, car elle augmente le risque de contamination. - Un étalement des produits d’amplification est observé sur le gel.
→ Interprétation : La matrice d’ARN est dégradée ou les puits du gel ont été surchargés.
→ Action : Améliorer l’intégrité de l’ARN par une manipulation soigneuse et une stabilisation immédiate après l’extraction, réduire la charge de la matrice et confirmer que les conditions d’électrophorèse sont appropriées. Si nécessaire, répéter la réaction avec une matrice purifiée et un nombre de cycles optimisé. - Les rendements d’amplification sont faibles ou les bandes sont faibles.
→ Interprétation : Une faible concentration initiale d’ARN, une transcription inverse inefficace ou la présence d’inhibiteurs de PCR (par ex., phénol ou éthanol résiduel) peuvent en être responsables.
→ Action : Mesurer la concentration et la pureté de l'ARN en utilisant les rapports A₂₆₀/A₂₃₀ — des valeurs faibles indiquent une contamination par des composés phénoliques ou guanidinium. S'assurer que les culots d'ARN sont correctement séchés avant remise en suspension. Si les inhibiteurs persistent, réextraire l'ARN ou effectuer des lavages supplémentaires à l'éthanol avant de répéter la RT-PCR.
Conclusion
Ce protocole offre un cadre robuste pour l'extraction d'ARN, la transcription inverse et la RT-PCR en point final, permettant une analyse fiable de l'expression génique, le clonage et le séquençage. En suivant ces étapes et en appliquant des mesures de contrôle qualité, les chercheurs peuvent obtenir des produits d'cDNA et de PCR de haute qualité pour des applications en biologie moléculaire.
Références:
-
Isolation of Total RNA from Yeast Cell Cultures by Manuel Ares, published in Cold Spring Harbor Protocols, 2012.
-
Reverse-Transcription PCR (RT-PCR) by Julia Bachman, Department of Neuroscience, Johns Hopkins University School of Medicine, published by Elsevier Inc., 2013.
-
The Polymerase Chain Reaction (PCR), qPCR, and RT-PCR by Mehdi Jalali, Justyna Zaborowska, and Morteza Jalali, University of Liverpool and University of Oxford, published by Elsevier Inc., 2017.